WHAT ARE MITOCHONDRIAL
DISEASES?
Just as some diseases are
named for the part of the body they affect (like heart disease), mitochondrial
diseases are so-named because they affect a specific part of the cells of which
our bodies are made. Specifically, mitochondrial diseases affect the
mitochondria — tiny energy factories found inside almost all our cells.
|
WHAT ARE
MITOCHONDRIAL DISEASES? |
|
|
|
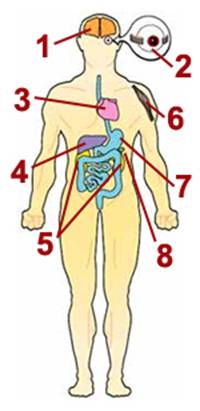
1. Nervous system
Seizures, spasms, developmental delays, deafness, dementia, stroke before
age 40, visual system defects, poor balance, problems with peripheral
nerves
2. Eyes
Drooping eyelids (ptosis), inability to move eyes from side to side
(external ophthalmoplegia), blindness (retinitis pigmentosa, optic
atrophy), cataracts
3. Heart
Cardiomyopathy (cardiac muscle weakness), conduction block
4. Liver
Liver failure (uncommon except in babies with mtDNA depletion syndrome)
5. Kidneys
Fanconi's syndrome (loss of essential metabolites in urine), myoglobinuria
6. Skeletal muscle
Muscle weakness, exercise intolerance, cramps
7. Digestive tract
Acid reflux, vomiting, chronic diarrhea, intestinal obstruction
8. Pancreas
Diabetes |
|
The main problems
associated with mitochondrial disease — low energy, free radical
production and lactic acidosis — can result in a variety of symptoms
in many different organs of the body. This diagram depicts common
symptoms of mitochondrial disease, of which most affected people
have a specific subset. Many of these symptoms are very treatable.
|
|
Mitochondria are
responsible for producing most of the energy that's needed for our cells to
function. In fact, they provide such an important source of energy that a
typical human cell contains hundreds of them. A mitochondrial disease can shut
down some or all the mitochondria, cutting off this essential energy supply.
Nearly all our cells rely
on mitochondria for a steady energy supply, so a mitochondrial disease can be a
multisystem disorder affecting more than one type of cell, tissue or organ. The
exact symptoms aren't the same for everyone, because a person with mitochondrial
disease can have a unique mixture of healthy and defective mitochondria, with a
unique distribution in the body.
Because muscle cells and
nerve cells have especially high energy needs, muscular and neurological
problems — such as muscle weakness, exercise intolerance, hearing loss, trouble
with balance and coordination, seizures and learning deficits — are common
features of mitochondrial disease. Other frequent complications include
cataracts, heart defects, diabetes and stunted growth. Usually, a person with a
mitochondrial disease has two or more of these conditions, some of which occur
together so regularly that they're grouped into syndromes.
A mitochondrial disease
that causes prominent muscular problems is called a mitochondrial myopathy (myo
means muscle, and pathos means disease), while a mitochondrial disease
that causes both prominent muscular and neurological problems is called a
mitochondrial encephalomyopathy (encephalo refers to the brain).
Despite their many
potential effects, mitochondrial diseases sometimes cause little disability.
Sometimes, a person has enough healthy mitochondria to compensate for the
defective ones. Also, because some symptoms of mitochondrial disease (such as
diabetes or heart arrhythmia) are common in the general population, there are
effective treatments for those symptoms (such as insulin or anti-arrhythmic
drugs).
This booklet describes
general causes, consequences and management of mitochondrial diseases, with an
emphasis on myopathies and encephalomyopathies and a close look at the most
common syndromes. These include:
·
Kearns-Sayre syndrome (KSS)
·
Leigh's syndrome
·
mitochondrial DNA depletion syndrome (MDS)
·
mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes (MELAS)
·
myoclonus epilepsy with ragged red fibers (MERRF)
·
mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
·
neuropathy, ataxia and retinitis pigmentosa (NARP)
·
Pearson syndrome
·
progressive external ophthalmoplegia (PEO)
WHAT CAUSES
MITOCHONDRIAL DISEASES?
First, mitochondrial
diseases aren't contagious, and they aren't caused by anything a person does.
They're caused by mutations, or changes, in genes — the cells' blueprints for
making proteins.
Genes are responsible for
building our bodies, and are passed from parents to children, along with any
mutations or defects they have. That means that mitochondrial diseases are
inheritable, although they often affect members of the same family in different
ways. (For more information about genetic mutations and mitochondrial disease,
see "Does
It Run in the Family?".)
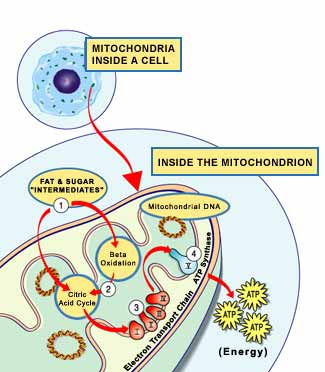
The genes involved in
mitochondrial disease normally make proteins that work inside the mitochondria.
Within each mitochondrion (singular of mitochondria), these proteins make up
part of an assembly line that uses fuel molecules derived from food to
manufacture the energy molecule ATP. This highly efficient manufacturing process
requires oxygen; outside the mitochondrion, there are less efficient ways of
producing ATP without oxygen.
Proteins at the beginning
of the mitochondrial assembly line act like cargo handlers, importing the fuel
molecules — sugars and fats — into the mitochondrion. Next, other proteins break
down the sugars and fats, extracting energy in the form of charged particles
called electrons.
Proteins toward the end of
the line — organized into five groups called complexes I, II, III, IV and
V — harness the energy from those electrons to make ATP. Complexes I
through IV shuttle the electrons down the line and are therefore called the
electron transport chain, and complex V actually churns out ATP, so it's
also called ATP synthase.
A deficiency in one or
more of these complexes is the typical cause of a mitochondrial disease. (In
fact, mitochondrial diseases are sometimes named for a specific deficiency, such
as complex I deficiency.)
When a cell is filled with
defective mitochondria, it not only becomes deprived of ATP — it can accumulate
a backlog of unused fuel molecules and oxygen, with potentially disastrous
effects.
Excess fuel molecules are
used to make ATP by inefficient means, which can generate potentially harmful
byproducts such as lactic acid. (This also occurs when a cell has an inadequate
oxygen supply, which can happen to muscle cells during strenuous exercise.) The
buildup of lactic acid in the blood — called lactic acidosis — is associated
with muscle fatigue, and might actually damage muscle and nerve tissue.
Meanwhile, unused oxygen
in the cell can be converted into destructive compounds called reactive oxygen
species. (These are the targets of so-called antioxidant drugs and vitamins.)
ATP derived from
mitochondria provides the main source of power for muscle cell contraction and
nerve cell firing. So, muscle cells and nerve cells are especially sensitive to
mitochondrial defects. The combined effects of energy deprivation and toxin
accumulation in these cells probably give rise to the main symptoms of
mitochondrial myopathies and encephalomyopathies.
WHAT HAPPENS TO SOMEONE
WITH A MITOCHONDRIAL DISEASE?
Myopathy
The main symptoms of
mitochondrial myopathy are muscle weakness and wasting, and exercise
intolerance. It's important to remember that the severity of any of these
symptoms varies greatly from one person to the next, even in the same family.
Weakness and wasting
usually are most prominent in muscles that control movements of the eyes and
eyelids. Two common consequences are the gradual paralysis of eye movements,
called progressive external ophthalmoplegia (PEO), and drooping of the
upper eyelids, called ptosis. Often, people automatically compensate for
PEO by moving their heads to look in different directions, and might not even
notice any visual problems. Ptosis is potentially more frustrating because it
can impair vision and also cause a listless expression, but it can be corrected
by surgery, or by using glasses that have a "ptosis crutch" to lift the upper
eyelids.
Mitochondrial myopathies
can also cause weakness and wasting in other muscles of the face and neck, which
can lead to slurred speech and difficulty with swallowing. In these instances,
speech therapy or changing the diet to easier-to-swallow foods can be useful.
Sometimes, people with mitochondrial myopathies experience loss of muscle
strength in the arms or legs, and might need braces or a wheelchair to get
around.
Exercise intolerance, also
called exertional fatigue, refers to unusual feelings of exhaustion brought on
by physical exertion. The degree of exercise intolerance varies greatly among
individuals. Some people might only have trouble with athletic activities like
jogging, while others might experience problems with everyday activities like
walking to the mailbox, or lifting a milk carton.
Sometimes, exercise
intolerance is associated with painful muscle cramps and/or injury-induced pain.
The cramps are actually sharp contractions that may seem to temporarily lock the
muscles, while the injury-induced pain is caused by a process of acute muscle
breakdown called
rhabdomyolysis. Cramps or rhabdomyolysis usually occur when someone with
exercise intolerance "overdoes it," and can happen during the overexertion or
several hours afterward.
Encephalomyopathy
A mitochondrial
encephalomyopathy typically includes some of the above-mentioned symptoms of
myopathy plus one or more neurological symptoms. Again, these symptoms show a
great deal of individual variability in both type and severity.
Hearing impairment,
migrainelike headaches and seizures are among the most common symptoms of
mitochondrial encephalomyopathy. In at least one syndrome, headaches and
seizures are accompanied by stroke (interruption of the brain's blood supply).
Fortunately, there are
good treatments for some of these conditions. Hearing impairment can be managed
using hearing aids and alternate forms of communication. Often, headaches can be
alleviated with medications and seizures can be prevented with drugs used for
epilepsy (anti-epileptics). Several drugs are currently under investigation for
treating stroke.
|

|
|
Occupational therapy is important for children with mitochondrial myopathy.
|
In addition to affecting
the musculature of the eye, a mitochondrial encephalomyopathy can affect the eye
itself and parts of the brain involved in vision. (For instance, cataracts —
thickening of the lenses that focus light in the eye — are a common symptom of
mitochondrial encephalomyopathy.) Compared to muscle problems, these effects are
more likely to cause serious visual impairment.
Often, mitochondrial
encephalomyopathy causes ataxia, or trouble with balance and coordination.
People with ataxia are usually prone to dizzy spells and falls, but can partly
avoid those problems through physical and occupational therapy, and the use of
supportive aids such as railings, a walker or — in severe cases — a wheelchair.
SPECIAL ISSUES IN
MITOCHONDRIAL MYOPATHIES AND ENCEPHALOMYOPATHIES
Respiratory Care
|

|
|
Mitochondrial myopathy can lead to respiratory problems that require support
from a ventilator.
|
Sometimes, these diseases
can cause significant weakness in the muscles that support breathing.
Also, mitochondrial
encephalomyopathies sometimes cause brain abnormalities that alter the brain's
control over breathing.
A person with mild
respiratory problems might require occasional supplemental oxygen, while someone
with more severe problems might require permanent support from a ventilator. If
you have a mitochondrial disorder, you should watch for signs of respiratory
insufficiency (such as shortness of breath or morning headaches), and have your
breathing checked regularly by a specialist.
Cardiac Care
Sometimes, mitochondrial
diseases directly affect the heart. In these cases, the usual cause is an
interruption in the rhythmic beating of the heart, called a conduction block.
Though dangerous, this condition is treatable with a pacemaker, which stimulates
normal beating of the heart. If you have a mitochondrial disorder, you may need
to have regular examinations by a cardiologist.
Other Potential Health
Issues
Some people with
mitochondrial disease experience serious kidney problems, gastrointestinal
problems and/or diabetes. Some of these problems are direct effects of
mitochondrial defects in the kidneys, digestive system or pancreas (in
diabetes), and others are indirect effects of mitochondrial defects in other
tissues.
For example,
rhabdomyolysis can lead to kidney problems by causing a protein called myoglobin
to leak from ruptured muscle cells and build up in the bloodstream. This
condition, called myoglobinuria, strains the kidneys' ability to filter waste
from the blood into urine, and can cause kidney damage.
Special Issues in
Children
Vision: Though PEO and
ptosis typically cause mild visual impairment in adults, they're potentially
more harmful in children with mitochondrial myopathies.
Because the development of
the brain is sensitive to childhood experiences, PEO or ptosis during childhood
can sometimes cause permanent damage to the brain's visual system. For this
reason, it's important for children with signs of PEO or ptosis to have their
vision checked by a specialist.
Developmental Delays: Due
to muscle weakness, brain abnormalities or a combination of both, children with
mitochondrial diseases may have difficulty developing certain skills. For
example, they might take an unusually long time to reach motor milestones (such
as sitting, crawling and walking). As they get older, they may be unable to get
around as easily as other children their age, and they might have speech
problems and/or learning disabilities. If your child is severely affected by
these problems, he or she may benefit from physical therapy, speech therapy and
possibly an individualized education program (IEP) at school.
|

|
|
Some children with mitochondrial myopathies experience developmental delays.
|
|

|
|
The
brain's visual system can be affected in children with mitochondrial
myopathy.
|
HOW ARE MITOCHONDRIAL
DISEASES TREATED?
While mitochondrial
myopathies and encephalomyopathies are relatively rare, some of their potential
manifestations are common in the general population. Consequently, those
complications (including heart problems, stroke, seizures, migraines, deafness
and diabetes) have highly effective treatments (including medications, dietary
modifications and lifestyle changes). (See "Special
Issues".)
It's fortunate that these
treatable symptoms are often the most life-threatening complications of
mitochondrial disease. With that in mind, people affected by mitochondrial
diseases can do a great deal to take care of themselves by monitoring their
health and scheduling regular medical exams.
Instead of focusing on
specific complications of mitochondrial disease, some newer, less-proven
treatments aim at fixing or bypassing the defective mitochondria. These
treatments are dietary supplements based on three natural substances involved in
ATP production in our cells.
One such substance,
creatine, normally acts as a reserve for ATP by forming a compound called
creatine phosphate. When a cell's demand for ATP exceeds the amount its
mitochondria can produce, creatine can release phosphate (the "P" in ATP) to
rapidly enhance the ATP supply. In fact, creatine phosphate (also called
phosphocreatine) typically provides the initial burst of ATP required for
strenuous muscle activity.
Another substance,
carnitine, generally improves the efficiency of ATP production by helping import
certain fuel molecules into mitochondria, and cleaning up some of the toxic
byproducts of ATP production. Carnitine is available as an over-the-counter
supplement called L-carnitine.
Finally, coenzyme Q10, or
coQ10, is a component of the electron transport chain, which uses oxygen to
manufacture ATP. Some mitochondrial diseases are caused by coQ10 deficiency, and
there's good evidence that coQ10 supplementation is beneficial in these cases.
Some doctors think that coQ10 supplementation might also alleviate other
mitochondrial diseases.
Creatine, L-carnitine and
coQ10 supplements are often combined into a "cocktail" for treating
mitochondrial disease. Although there's little scientific evidence that this
treatment works, many people with mitochondrial disease have reported modest
benefits. At the very least, there appear to be almost no harmful side effects
to the three supplements when they're taken in moderation, but you should
consult your doctor or MDA clinic director before taking any of them.
WHAT SYNDROMES OCCUR
WITH MITOCHONDRIAL DISEASE?
Note: Typically, these
syndromes are inherited in either a maternal pattern ( )
or a so-called Mendelian pattern (
)
or a so-called Mendelian pattern ( ),
and/or they're sporadic (
),
and/or they're sporadic ( ),
which means occurring with no family history. For more information about
inheritance, see "Does
It Run in the Family?"
),
which means occurring with no family history. For more information about
inheritance, see "Does
It Run in the Family?"
KSS: Kearns-Sayre
syndrome
Onset: before age 20
Features: This disorder is defined by PEO (usually as the initial
symptom) and pigmentary retinopathy, a "salt-and-pepper" pigmentation in the
retina that can affect vision, but often leaves it intact. Other common symptoms
include conduction block (in the heart) and ataxia. Less typical symptoms are
mental retardation or deterioration, delayed sexual maturation and short
stature.
Leigh's syndrome:
subacute necrotizing encephalomyopathy (MILS
= maternally inherited Leigh's syndrome)
Onset: infancy
Features: Leigh's syndrome causes brain abnormalities that can result in
ataxia, seizures, impaired vision and hearing, developmental delays and altered
control over breathing.
It also causes muscle weakness, with prominent effects on swallowing, speech and
eye movements.
MDS: mitochondrial DNA
depletion syndrome
Onset: infancy
Features: This disorder typically causes muscle weakness and/or liver
failure, and more rarely, brain abnormalities. "Floppiness," feeding
difficulties, and developmental delays are common symptoms; PEO and seizures are
less common.
MELAS: mitochondrial
encephalomyopathy, lactic acidosis and strokelike episodes
Onset: childhood to early adulthood
Features: MELAS causes recurrent strokes in the brain, which manifest as
migrainelike headaches, vomiting and (less often) seizures, and can lead to
permanent brain damage. Other common symptoms include PEO, general muscle
weakness, exercise intolerance, hearing loss, diabetes and short stature.
MERRF: myoclonus
epilepsy with ragged red fibers
Onset: late childhood to adolescence
Features: The most prominent symptoms are myoclonus (muscle spasms),
seizures, ataxia and muscle weakness. The disease can also cause hearing
impairment and short stature.
MNGIE: mitochondrial
neurogastrointestinal encephalomyopathy
Onset: usually before age 20
Features: This disorder causes PEO, ptosis, limb weakness and
gastrointestinal (digestive) problems, including chronic diarrhea and abdominal
pain. Another common symptom is peripheral neuropathy (a malfunction of the
nerves that can lead to sensory impairment and muscle weakness).
NARP: neuropathy,
ataxia and retinitis pigmentosa
Onset: infancy to adulthood
Features: NARP causes neuropathy (see above), ataxia and retinitis
pigmentosa (degeneration of the retina in the eye, with resulting loss of
vision). It can also cause developmental delay, seizures and dementia.
Pearson syndrome
Onset: infancy
Features: This syndrome causes severe anemia and malfunction of the
pancreas. Children who survive the disease usually go on to develop KSS.
PEO: Progressive
external ophthalmoplegia
Onset: Usually in adolescence or early adulthood
Features: As noted above, PEO is often a symptom of mitochondrial
disease, but sometimes it stands out as a distinct syndrome. Often, it's
associated with exercise intolerance.
HOW ARE MITOCHONDRIAL
DISEASES DIAGNOSED?
None of the hallmark
symptoms of mitochondrial disease — muscle weakness, exercise intolerance,
hearing impairment, ataxia, seizures, learning disabilities, cataracts, heart
defects, diabetes and stunted growth — are unique to mitochondrial disease.
However, a combination of three or more of these symptoms in one person strongly
points to mitochondrial disease, especially when the symptoms involve more than
one organ system.
To evaluate the extent of
these symptoms, a physician usually begins by taking the patient's personal
history, and then proceeds with physical and neurological exams.
|
DIAGNOSTIC TESTS IN MITOCHONDRIAL DISEASES
|
|
Type |
Test |
What It
Shows |
|
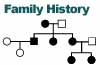
|
Clinical exam or oral history of family members |
Can sometimes indicate inheritance pattern by noting "soft signs" in
unaffected relatives. These include deafness, short stature, migraine
headaches and PEO. |
|
|
1.
Histochemistry
2.
Immuno-histochemistry
3. Biochemistry
4. Electron
microscopy |
1.
Detects abnormal proliferation of mitochondria and deficiencies in
cytochrome C oxidase (COX, which is complex IV in the electron transport
chain).
2. Detects presence
or absence of specific proteins. Can rule out other diseases or confirm
loss of electron transport chain proteins.
3. Measures
activities of specific enzymes. A special test called polagraphy measures
oxygen consumption in mitochondria.
4. May confirm
abnormal appearance of mitochondria. Not used much today. |
|
|
1.
Lactate and pyruvate levels
2. Serum creatine
kinase |
1.
If elevated, may indicate deficiency in electron transport chain; abnormal
ratios of the two may help identify the part of the chain that is blocked.
2. May be slightly
elevated in mitochondrial disease but usually only high in cases of
mitochondrial DNA depletion. |
|

|
1.
Known mutations
2. Rare or unknown
mutations |
1.
Uses blood sample or muscle sample to screen for known mutations, looking
for common mutations first.
2. Can also look for
rare or unknown mutations but may require samples from family members;
this is more expensive and time-consuming. |
The physical exam
typically includes tests of strength and endurance, such as an exercise test,
which can involve activities like repeatedly making a fist, or climbing up and
down a small flight of stairs. The neurological exam can include tests of
reflexes, vision, speech and basic cognitive (thinking) skills.
Depending on information
found during the personal history and exams, the physician might proceed with
more specialized tests that can detect abnormalities in muscles, brain and other
organs.
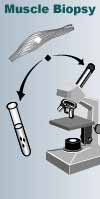
The most important of
these tests is the muscle biopsy, which involves removing a small sample of
muscle tissue to examine. When treated with a dye that stains mitochondria red,
muscles affected by mitochondrial disease often show ragged red fibers — muscle
cells (fibers) that have accumulated mitochondria. Other stains can detect the
absence of essential mitochondrial enzymes in the muscle. It's also possible to
extract mitochondrial proteins from the muscle and measure their activity.
In addition to the muscle
biopsy, noninvasive techniques can be used to examine muscle without taking a
tissue sample. For instance, a technique called muscle phosphorus magnetic
resonance spectroscopy (MRS) can measure levels of phosphocreatine and ATP
(which are often depleted in muscles affected by mitochondrial disease). Also, a
CT scan or magnetic resonance imaging (MRI) are tools for visualizing the
overall structure of muscles.
CT scans and MRI can also
be used to visually inspect the brain for signs of damage, and surface
electrodes placed on the scalp can be used to produce a record of the brain's
activity called an electroencephalogram (EEG).
A blood test might be
ordered so the lab can look for a buildup of lactate or unused fuel molecules in
the blood or cerebrospinal fluid (fluid that bathes the brain and spinal cord).
Similar techniques might
be used to examine the functions of other organs and tissues in the body. For
example, an electrocardiogram (EKG) can monitor the heart's activity, and a
blood test can detect signs of kidney malfunction.
Finally, a genetic test
can determine whether someone has a genetic mutation that causes mitochondrial
disease. Ideally, the test is done using genetic material extracted from blood
and from a muscle biopsy. It's important to realize that, although a positive
test result can confirm diagnosis, a negative test result isn't necessarily
meaningful.
DOES IT RUN IN THE
FAMILY?
Often, a mitochondrial
disease can be difficult to trace through a family tree. But since they're
caused by defective genes, mitochondrial diseases do run in families.
To understand how
mitochondrial diseases are passed on through families, it's important to know
that there are two types of genes essential to mitochondria. The first type is
housed within the nucleus — the part of our cells that contains most of our
genetic material, or DNA. The second type resides exclusively within DNA
contained inside the mitochondria themselves.
Mutations in either
nuclear DNA (nDNA) or mitochondrial DNA (mtDNA) can cause mitochondrial disease.
Most nDNA (along with any
mutations it has) is inherited in a Mendelian pattern, loosely meaning that one
copy of each gene comes from each parent. Also, most mitochondrial diseases
caused by nDNA mutations (including Leigh's syndrome, MNGIE and even MDS) are
autosomal recessive, meaning that it takes mutations in both copies of a gene to
cause disease.
Unlike nDNA, mtDNA passes
only from mother to child. That's because during conception, when the sperm
fuses with the egg, the sperm's mitochondria — and its mtDNA — are destroyed.
Thus, mitochondrial diseases caused by mtDNA mutations are unique because
they're inherited in a maternal pattern (see illustration).
Another unique feature of
mtDNA diseases arises from the fact that a typical human cell — including the
egg cell — contains only one nucleus, but hundreds of mitochondria. The upshot
is that a single cell can contain both mutant mitochondria and normal
mitochondria, and the balance between the two will determine the cell's health.
This helps explain why the
symptoms of mitochondrial disease can vary so much from person to person, even
within the same family.
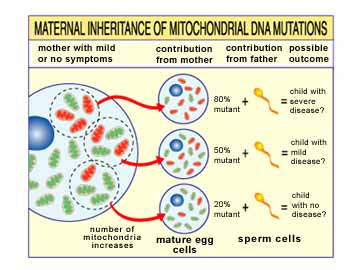
Imagine that a woman's egg
cells (and other cells in her body) contain both normal and mutant mitochondria,
and that some have just a few mutant mitochondria, while others have many. A
child conceived from a "mostly healthy" egg cell probably won't develop disease,
and a child conceived from a "mostly mutant" egg cell probably will.
Also, the woman may or may
not have symptoms of mitochondrial disease herself.
The risk of passing on a
mitochondrial disease to your children depends on many factors, including
whether the disease is caused by mutations in nDNA or mtDNA.
A good way to find out
more about these risks is to talk to a doctor or genetic counselor at your local
MDA clinic. Also, see MDA's pamphlet, "Genetics
and Neuromuscular Diseases."
MDA'S SEARCH FOR
TREATMENTS AND CURES
With MDA's support,
scientists continue to make significant progress in their quest to fully
understand and treat mitochondrial diseases.
Because mitochondrial
diseases can cause very diverse symptoms, they can be challenging to diagnose,
and have historically been misdiagnosed as other diseases. MDA-funded scientists
have helped improve diagnosis by carefully identifying the hallmark features of
mitochondrial disease.
In an ongoing effort, MDA-funded
scientists also have identified many of the genetic defects that cause
mitochondrial diseases. They've used knowledge of those genetic defects to
create animal models of mitochondrial disease, which can be used to investigate
potential treatments. They've also designed genetic tests that allow accurate
diagnosis of mitochondrial defects and provide valuable information for family
planning.
Perhaps most important,
knowing the genetic defects that cause mitochondrial disease opens up the
possibility of one day repairing those defects via gene therapy.
While some of MDA's
scientists pursue gene therapy for mitochondrial diseases, others are conducting
clinical trials to evaluate the benefits of dietary supplements (such as
creatine, carnitine and coQ10) and certain drugs (such as dichloroacetate, which
can reduce lactic acidosis).
For people who have
diseases caused by mtDNA mutations, MDA-funded scientists are working on several
novel treatment strategies. For example, mtDNA isn't readily accessible to
conventional gene therapy (because it's trapped inside mitochondria), so
scientists are developing new gene therapy techniques to overcome that obstacle.
Also, some scientists are investigating drug treatments and certain types of
exercise to increase cellular levels of normal mtDNA relative to mutant mtDNA.
|
Questions |
|
The
Children’s Mitochondrial Disease Network is the only parental and
professional based registered organization Within the
UK
specializing in the complexities of Mitochondrial and sssociated disorders.
What are
Mitochondria?
Mitochondria are
small complex structures, which exist in every cell of the body (except red
blood cells). The mitochondrion has been called the ‘powerhouse’ of the
cell, as these tiny structures produce most of the energy, which we all need
to grow and live. Those organs in the body, which require a lot of energy to
work properly, are particularly dependent on well functioning mitochondria.
The most energy dependent organs are the brain, heart, skeletal muscle,
kidney, endocrine glands and bone marrow and these are the organ systems
commonly affected in mitochondrial diseases.
There are from one
to several hundred mitochondria in each cell and each mitochondrion contains
the complex molecules necessary to carry our energy making chemical
reactions. Mitochondria perform many functions necessary for cell metabolism
but the energy producing pathways are the most important. These pathways
allow us to break down carbohydrate, fat and oxygen to live. Electrons from
these food molecules are passed down a series of complex molecules called
the electron transport chain. The final molecule in the chain, cytochrome
oxidase, passes the electrons to oxygen
One unique feature
of mitochondria is that they have there own DNA molecules, mitochondrial
DNA, which carries the genes containing the genetic message for several
critical components of the electron transport chain.
What is a
Mitochondrial Disease?
When enough
mitochondria are not working correctly a disease may result. Mitochondrial
diseases often involve the brain because of the tremendous energy
requirements of the brain cells. Mitochondrial diseases are very variable in
their features so called clinical heterogeneity.
The variability
results from the fact that different organ systems contain different amounts
of diseased mitochondria and only those tissues with a high percentage of
diseased mitochondria will be functional impaired. Mitochondrial diseases
are whole body diseases but the exact features of the disease vary from one
patient to another. Some patients will have predominately brain disease or
nerve disease. Others will have muscle disease (mitochondrial myopathy),
cardiac disease (cardiomyopathies), endocrine, renal or bone marrow disease
or a mixture of these and or other features.
Many mitochondrial
diseases result in the accumulation of organic acids in the body. These are
usually normal metabolic intermediates but when present in excess, the
acidosis itself may be damaging or even life-threatening. Lactic acid
accumulation is a common problem in mitochondrial diseases.
We used to think of
mitochondrial diseases as rare childhood disorders. Recently it has been
discovered that many commoner disorders such as diabetes and ischemic heart
disease have, in some cases, a mitochondrial basis. Also, diseases of aging
such as Parkinson’s disease and Alzheimer’s disease may result in part from
mitochondrial failure (The role that mitochondrial abnormalities play in the
cause of these diseases remains to be established). In fact, the aging
process itself may be due to a lifetime of damage to mitochondria through
oxidative stress and accumulated damage to mitochondrial proteins and
mitochondrial DNA.
Genetics of
Mitochondrial Diseases?
Some mitochondrial
diseases are clearly inherited and those involving mitochondrial DNA may be
inherited through the maternal side of the family as almost all mitochondria
come from the mother. Most inherited mitochondrial diseases however are so
called nuclear DNA defects with inheritance from either the mother or
father, or in most cases both. This latter inheritance |
How are Mitochondrial
Diseases diagnosed?
Because of the
multiple organ systems involved and the variation in the age of onset,
mitochondrial diseases may be difficult to recognize. Even within the same
family the same disease may affect individuals differently. A severe
childhood disease such as Leigh’s syndrome may occur in the same family with
later onset adult neurodegenerative disease. In some families mitochondrial
myopathy has found some members with deafness and diabetes in others strokes
along with a mixture of other symptoms. As well as the history and physical
examination, blood and urine specialized tests together with brain CT or MRI
scanning and skin and muscle biopsy are often needed to make a diagnosis.
Patients should be referred as soon as possible to a specialist centre with
expertise in metabolic and mitochondrial diseases.
What Treatments
are available? (1)
In the New
Millennium treatments for mitochondrial diseases are not very effective.
Some effects of these diseases can be treated such as cardiac arrhythmia,
seizure disorders, renal bicarbonate loss and hypoglycaemia.
When lactic acid
accumulation seems to be a major problem an experimental drug Dichloroacete
DCA, will lower the lactic acid. Although conclusive evidence of efficacy is
not yet available, most doctors working with mitochondrial diseases treat
their patients with cofactors and vitamins, which are thought to help
impaired metabolic pathways. These treatments include combinations of
Coenzyme Q10, L-Carnitine, Niacin, Thiamine, Biotin and Riboflavin. Special
diets can be helpful.
Some patient’s
benefit by high fat diets with restriction of simple carbohydrates. Fructose
restriction may help.
Other patients need
high carbohydrate intake with particular supplementation of complex
carbohydrates such as uncooked cornstarch.
Only with a through
medical evaluation, best carried out in a centre specializing in metabolic &
mitochondrial diseases, can the optimal treatment regime for each patient be
chosen.
What does the
future hold? (1a)
There is no
convincing evidence to date of any clear benefit of drug therapies in most
archetypal mitochondrial disorders or those neurodegenerative conditions
with evidence of mitochondrial dysfunction, and therefore attention has
turned to the development of genetic therapies and the possibility of Neuro
protection.
New horizons and
hopes may lie with genetic strategies. Techniques for manipulating the
mitochondrial genome are now being investigated. Whereas nuclear
manipulation would necessitate treatment for life, manipulation of the
mitochondrial genome would result in a one-off treatment thus providing a
pattern is termed autosomal recessive and in this case the risk of
reoccurrence in a sibling is 25% or one in 1 in 4. Most childhood onset
mitochondrial diseases are inherited although in some cases the affected
child seems to be the only affected family member.
Diseases resulting
from mitochondrial deletions of large parts of the mitochondrial DNA
molecule are usually sporadic without other affected family members. Genetic
counseling is complex for mitochondrial diseases.
Pre-natal testing
is only available for a few disorders.
“CURE” for
Mitochondrial Disorders.
None Known
at this time!
|